
With the introduction of the Medical Device Regulation 2017/745 (MDR), the regulatory requirements for the clinical documentation of medical devices have become more challenging. Uncertainties persist about how to precisely follow the newly introduced rules, worsened by variations in how different reviewers and regulatory bodies assess them. Our experience indicates that a growing emphasis is placed on a careful and early planning of the clinical evaluation strategy.
The requirements for performing clinical evaluations under the MDR are mainly defined in MDR Article 61 and Annex XIV Part A. However, the MDR does not provide detailed instructions for the actual execution of clinical evaluations and leaves quite some room for interpretations. Despite the joint efforts to provide more explicit guidelines for both manufacturers and notified bodies in various MDCG guidance documents, the use of vague terminology leads to heterogeneous clinical evaluation reviews during conformity assessment.
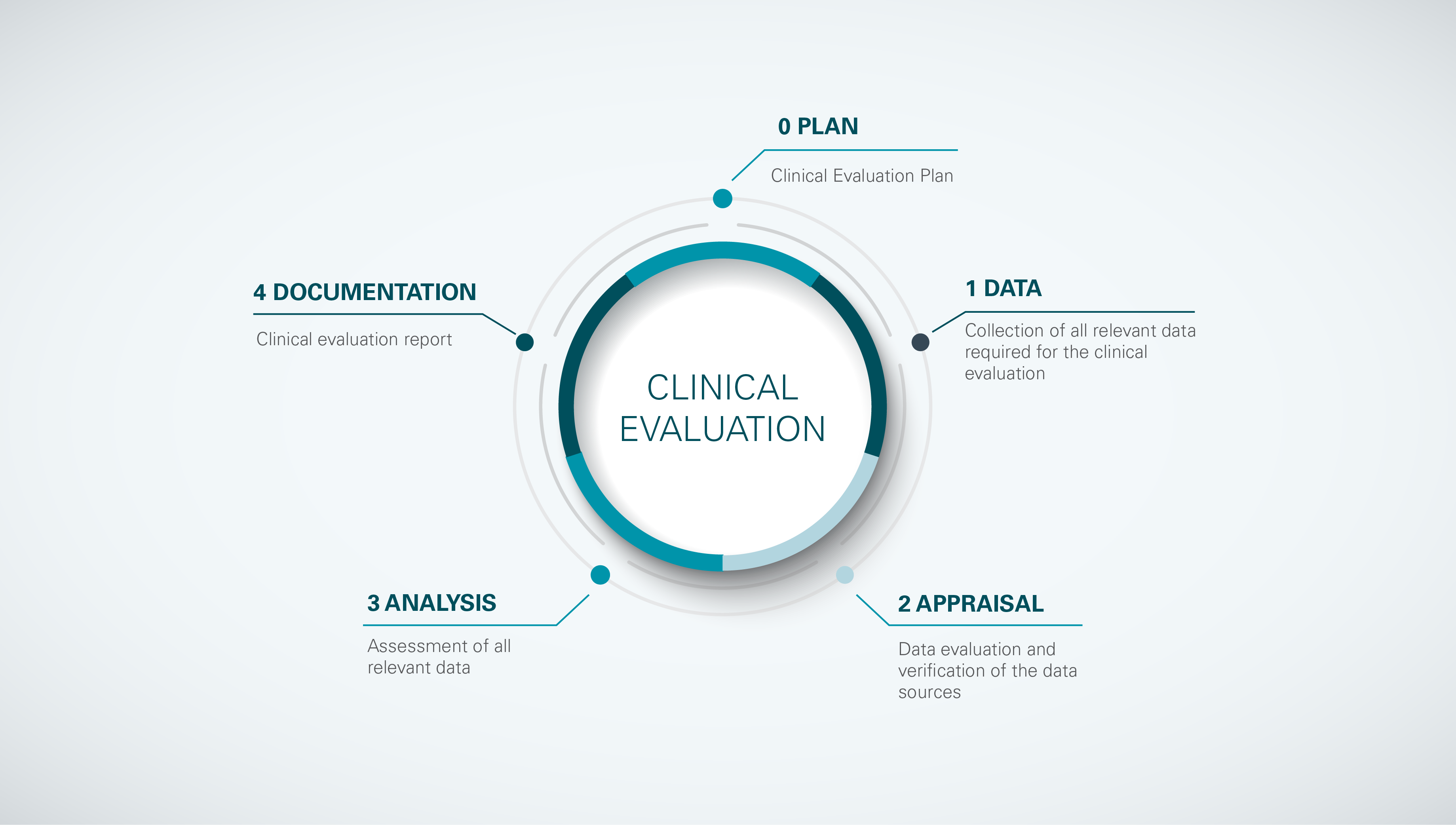 The 5 stages of the clinical evaluation process according to MEDDEV 2.7/1 Rev.4.
The 5 stages of the clinical evaluation process according to MEDDEV 2.7/1 Rev.4.
Clinical evaluation should be perceived as a continuous cycle rather than a linear process. By considering aspects from stages 1 to 4 already during the planning phase (stage 0), manufacturers can avoid unpleasant surprises in later phases of product development. In alignment with Stage 0 for performing clinical evaluations according to MEDDEV 2.7/1 Revision 4, the MDR requires that manufacturers establish a Clinical Evaluation Plan and update it throughout the device life cycle. This planning document is often regarded as an unnecessary addendum to the Clinical Evaluation Report required as a formality to receive CE-marking. However, our extensive experience and feedback from different notified bodies show that following a sound clinical evaluation strategy has become an important aspect during conformity assessment. This strategy is outlined in the Clinical Evaluation Plan. It identifies the required level of clinical evidence for demonstrating conformity with clinically relevant General Safety and Performance Requirements, as well as the methods for clinical data collection and evaluation.
Thorough planning of the clinical evaluation strategy also includes an evaluation of the state of the art for the concerned medical device, which provides valuable inputs for other aspects of device development, including risk management and preclinical testing. An early assessment of the state of the art allows for the identification of safety and performance indicators relevant to the concerned device, as well as appropriate test methods to demonstrate that predefined acceptance criteria are met. If this step is performed carefully and correctly, unpleasant surprises at later stages of product development can be avoided and project delays e.g. due to a need for additional data can be prevented.
The Clinical Evaluation Plan is therefore an extremely valuable document for the manufacturer and should be regarded as a helpful guide for all (pre-)clinical activities throughout the entire product life cycle. Following a tailored clinical strategy is especially important for successful and efficient product development, allowing a manufacturer to identify project risks and potential roadblocks. We highly recommend starting the clinical evaluation planning during the early device development phase, and this is in line with our recent experience with feedbacks from notified bodies.
Important aspects of clinical evaluation planning are:
- Identifying clinically relevant aspects of product design that need to be considered during development.
- Assessing the necessity of performing comparative preclinical or bench testing.
- Defining all relevant outcome parameters for product safety, performance, and clinical benefits for all proposed indications.
- (Re)assessing the evolving state of the art and benchmarks to identify the acceptance criteria that need to be achieved.
- Evaluating device-specific aspects and the suitability of potential strategies that might affect the required level of clinical evidence and the necessity for collecting own clinical data, including:
- device risk class;
- previous marketing under MDD or AIMDD (legacy devices);
- claiming of equivalence;
- applicability of MDR Article 61(10) for justification that clinical data is not appropriate;
- meeting of requirements for well-established technologies/standard-of-care devices set out in MDCG 2020-6;
- Providing input on the design of pre-market clinical investigations or post-market clinical follow-up activities.
Investing time into the early planning of the clinical strategy leads to various benefits for the manufacturer during development and the entire product life cycle, and ultimately contributes to a flawless conformity assessment.
Kathrin Abegg, DVM PhD
Team Leader Medical Writing ISS AG
Ronja Weber, PhD
Medical Writer ISS AG