
As everyone is now aware, post-market clinical evidence for medical devices sold in Europe must be regularly collected and assessed.
The EU Medical Device Regulation 2017/745 (MDR) demands an increased level of scrutiny of medical devices by notified bodies to guarantee their safety and performance for the entire lifecycle of the product. Sufficient clinical evidence of an acceptable quantity and quality is required to continually assess whether the device is safe and achieves its intended purpose.
But what exactly do they mean by sufficient?
Clinical evidence is defined in the MDR as clinical data and clinical evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer. Despite the extensive MDR document as well as multiple MDCG Guidance documents, understanding the meaning of sufficient clinical evidence represents a significant challenge to compliance with the regulation. Rather than confront the data collection, workaround tactics such as reducing the indications for use or removing devices from the EU market have been implemented.
Avoiding the topic of clinical evidence is not the solution, and the process doesn’t have to be as overwhelming as anticipated.
While third-party data (i.e., vigilance data and literature reviews) is part of the assessment, relying solely upon that data as evidence may increase the chance of falling into the trap of poor quality. The best way to ensure the quality of the data is to actively collect it. However, do not assume that a long, drawn-out clinical investigation is always needed. There are multiple acceptable methods of collecting clinical data that don’t need to be as costly and time-consuming as a clinical study. Post-market clinical follow-up (PMCF) activities are meant to address specific safety and performance questions. The two main categories are studies and surveys. The table below shows the two categories and the types of activities that correspond to each.
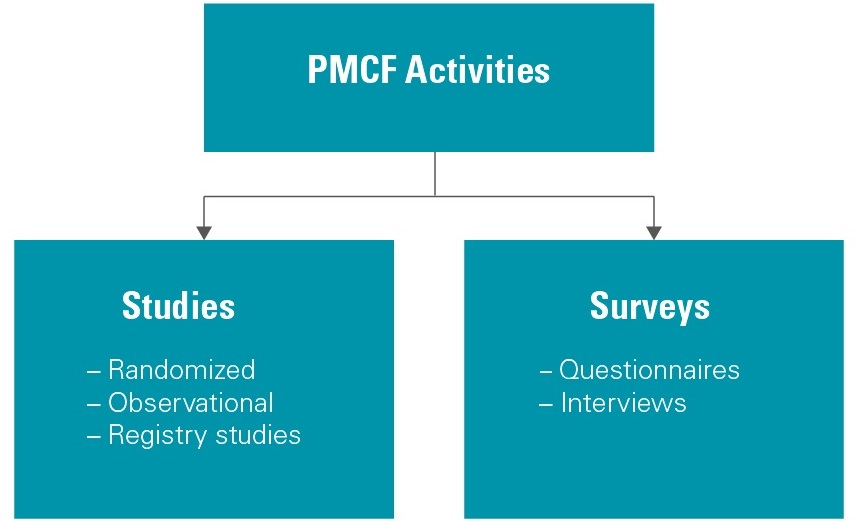
Figure 1. PMCF activity categories and methods
Post-market activities allow for the collection of data from a medical device as used during standard care with its intended use and in its intended environment.
During its lifetime, the use of a medical device generates real-world data (RWD) and analysing that data allows for the compilation of real-world evidence (RWE) related to the risks and benefits of that device. RWD can be collected from various sources including health records, claims and billing activities, product and disease registries, and patient-generated data (i.e., mobile devices that inform on health status). Study designs used to collect this type of data can range from randomised trials to observational prospective studies, and retrospective chart reviews and will vary depending on the number of subjects required. Surveys are another method of collecting data that involve questionnaires and interviews conducted with either patients or clinicians that will often focus on the usability, user experience, and opportunities for improvement of a device.
Underpinning both categories is the intent to gather quality data by conducting the activities using a planned and systematic approach.
Scientific design and methods shall be applied to all activities to ensure the integrity and applicability of the results. The two categories diverge at the level of subjectivity. Surveys, although carefully planned, will still have an increased level of subjectivity, and offer less control over the raw data.
The type of activity selected will depend on the PMCF objective – to determine residual or unknown risks, to ensure that the intended use remains valid, or that marketing claims still apply.* A sound rationale for the appropriateness of the chosen methods to achieve those objectives will justify the ability to make a qualified assessment of the product from the resulting data. The quantity of data required is related to the existing amount of data for the product. A clinical evaluation strategy for the lifecycle of the product created during the initial stages of development will manage the data collection activities from the start. Furthermore, periodic reviews and updates regarding those activities will ensure a consistent and evolving process rather than a chaotic search to find clinical evidence at the last minute.
Be proactive rather than reactive.
To relieve the burden of post-market clinical evidence, invest time and effort into the organisation of data collection during the initial development of the product. Likewise, for legacy products, take the time to perform an extensive review and gap analysis of the portfolio. Planning the appropriate PMCF activities rather than depending on existing general data or resorting to a clinical investigation out of desperation will ensure a quality assessment of the device without an excessive amount of effort. Re-evaluate the product on a regular basis to identify new gaps in the clinical data and plan how to address them, thus, removing the uncertainty of how much and what type of data is sufficient to fill those gaps.
*Note that testing a CE-marked device outside the scope of its intended purpose does not fall under PMCF activities - articles 62-81 of MDR will apply.
Julie Tantau, AuD
Team Leader, CRO